Efficacy
EFFICACY
EFFICACY IN THE 1-YEAR PIVOTAL TRIAL1
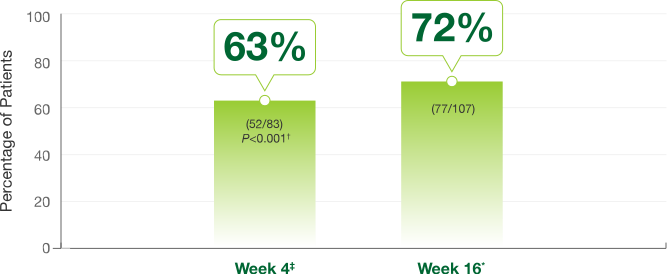
Primary endpoint at Week 4
- 63% of Somatuline Depot patients (52/83) achieved >50% decrease of growth hormones vs 0% of placebo patients (0/25) (P<0.001)
Efficacy achieved in the first 16 weeks was maintained through 52 weeks*1,2
Patients experienced a >50% decrease in GH across all doses
- 82% of patients experienced >50% reduction in GH at Week 52 (81/99)*
Study design: In a 1-year study including a 4-week, double-blind, placebo-controlled phase (N=107); After Week 4, all patients received active drug and entered a 16-week, single-blind, fixed-dose phase (N=105) and a 32-week, open-label, dose-titration phase (N=99) injections of 60, 90, or 120 mg were given at 4-week intervals. During the dose-titration phase of the study, the dose was titrated twice, if needed, according to individual GH and IGF-1 levels.
*Week 16 and Week 52 data were secondary endpoints in the pivotal trial.2
†P value is vs placebo.
‡Week 4 data were a primary endpoint in the pivotal trial.2
IGF-1 Normalization Responders§
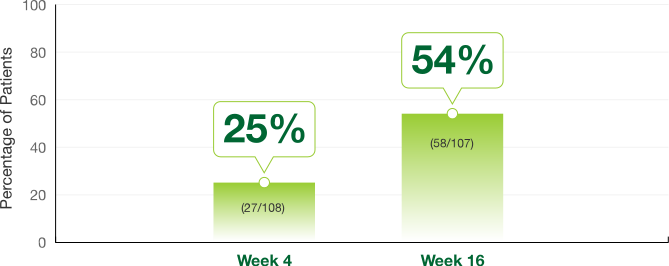
- 59% of patients experienced IGF-1 normalization at Week 52 (57/99)§
§IGF-1 data analyses were secondary endpoints of the pivotal trial.
EXTENDED DOSING INTERVALS (EDIs)1,3
Somatuline Depot was the first somatostatin analog (SSA) to offer FDA-approved EDI for controlled¶ patients†1,3
In an open-label, uncontrolled, multicenter, phase 3 trial3
Biochemical control was maintained with 120-mg dosing administered once every 6 or 8 weeks3
¶Controlled is defined as GH level from >1.0 ng/mL to ≤2.5 ng/mL, normalized IGF-1 level, and satisfactory management of clinical symptoms as determined by the healthcare professional.
†Patients who are controlled with Somatuline Depot 60 mg or 90 mg administered every 4 weeks can be considered for treatment with 120 mg administered every 6 or 8 weeks. GH and IGF-1 levels should be obtained 6 weeks after this change in dosing regimen to evaluate persistence of patient response. Continued monitoring of patients’ response with dose adjustments for biochemical and clinical symptom control, as necessary, is recommended.
Study design: In an open-label, comparative, multicenter, phase 3 trial, Somatuline Depot (lanreotide) Injection 120 mg was administered every 4, 6, or 8 weeks in patients previously receiving lanreotide microparticles every 5-7, 8-11, or 12-16 days, respectively. Of patients whose levels were controlled (GH ≤2.5 ng/mL and normalized IGF-1) when switched to extended dosing intervals (n=32), 5 out of 6 remained controlled after 3 injections at 6-week intervals and 23 out of 26 remained controlled after 3 injections at 8-week intervals.
Pharmacokinetic Profile
PHARMACOKINETIC PROFILE
PHARMACOKINETIC (PK) PROFILE DURING EXTENDED DOSING INTERVALS (EDIs)3
Cmin of lanreotide after a single deep subcutaneous injection in healthy volunteers (mean ± SD)
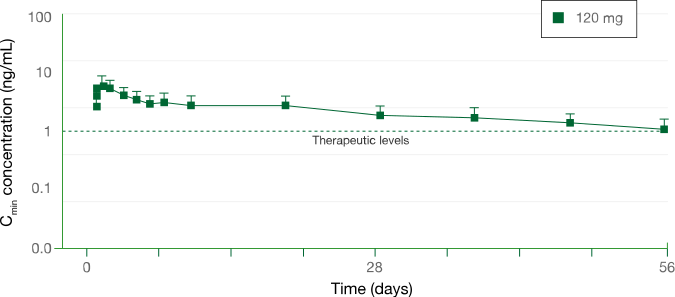
Study design: In a phase 1, single-center, open-label, randomized, parallel-group study, the pharmacokinetic profile of a single injection of lanreotide was assessed in healthy volunteers at a dose of 120 mg (n=12) through 56 days (8 weeks).
In patients treated with Somatuline Depot 120 mg:
- Serum concentration (Cmin) through 6- and 8-week dosing intervals3
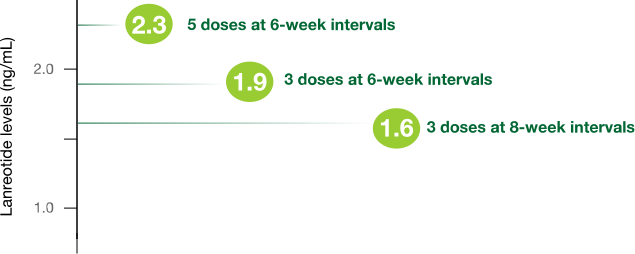
Study design: In open-label, comparative, multicenter, phase 3 trials, eligible patients who responded to SSAs received 3 to 5 injections of Somatuline Depot 120 mg. Somatuline Depot 120 mg was injected every 4, 6, or 8 weeks in patients previously receiving lanreotide microparticles every 5-7, 8-11, or 12-16 days, respectively. There was no washout period or dose titration.